Molecular Modeling of Mutant Caspases: A Structural Analysis of Conformational Changes
This study uses molecular modeling to analyze structural changes in mutant caspases. Molecular dynamics simulations reveal distant conformational alterations linked to mutations, offering insights into enzyme activity.
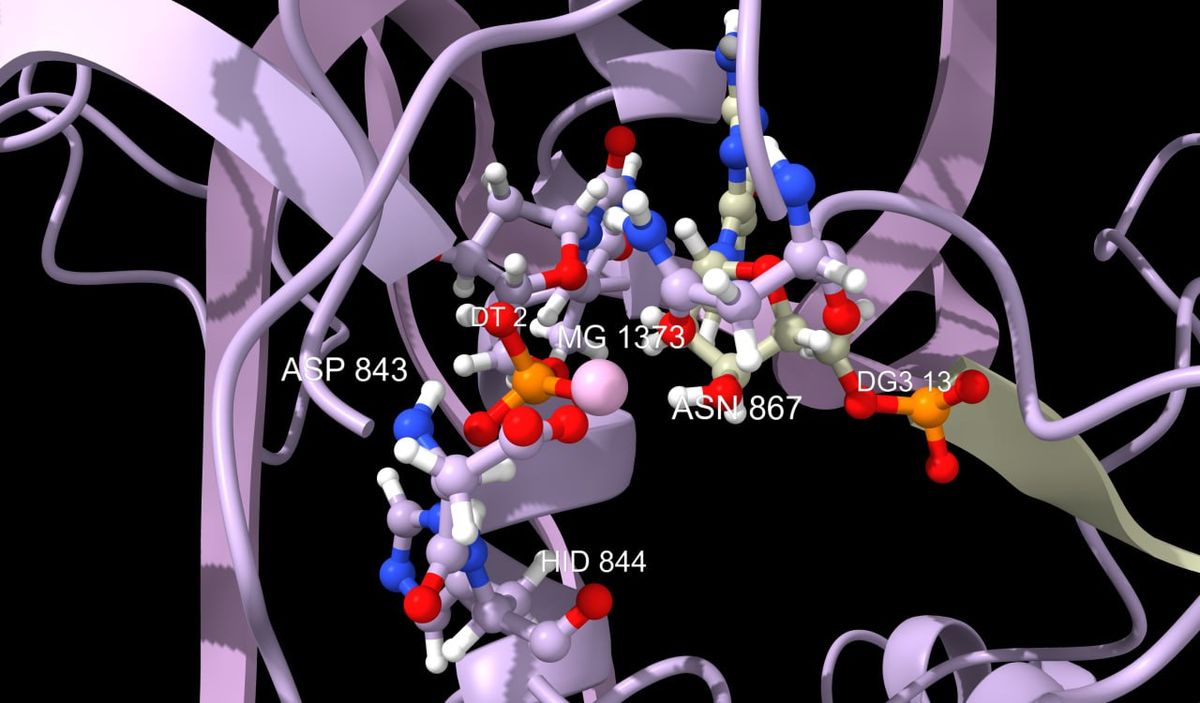
The discovery of the genome editing mechanism using caspase proteins, capable of recognizing specific DNA segments and cutting them out, is a promising tool for biotechnology. Unfortunately, existing caspases are far from perfect and do not yet provide enough accuracy for their widespread use in medicine and biotechnology. In this regard, work is being carried out to search for mutations that can enhance their effectiveness.
Specialists from our laboratory took part in one of the experimental works, where the activity of several Cas9 mutation variants was studied. The authors made assumptions about the reasons for the changes in the activity of some mutant Cas9, based on the results of the experiment, as well as a review of the existing literature at that time. The essence of the case that was brought to us was that the reviewer suggested using molecular dynamics methods to delve deeper into the issue and provide additional support for the experimental results.
In this article, we will not delve into technological aspects, but will focus on some significant results that were obtained using modeling methods.
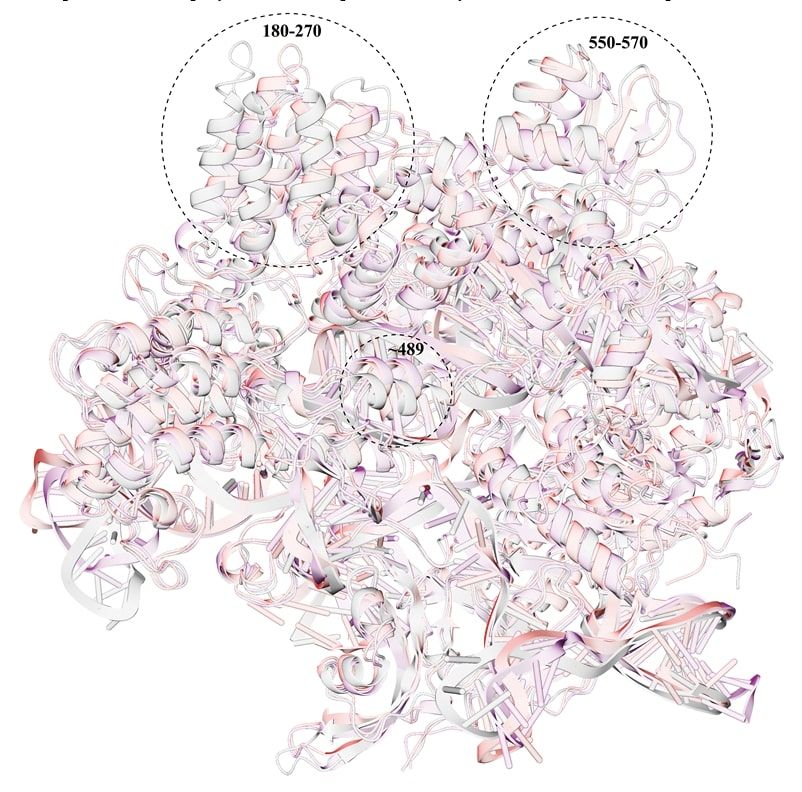
After conducting multistage MD simulations, equilibrium geometries of the Cas9 mutants were obtained. Attention is drawn to the fact that strong distortions of geometries are observed in regions that are located quite far from the mutation points. Judging by the direction of the chains, the group of mutations D1135E+D147Y+P411T causes greater compaction of the enzyme.
In the key regions of mutations (1135, 1206, 147, and 411), an analysis of hydrogen bonds was carried out, and the geometries were compared with the corresponding region of the wild-type Cas9. The results are presented in Figures 2 and 3.
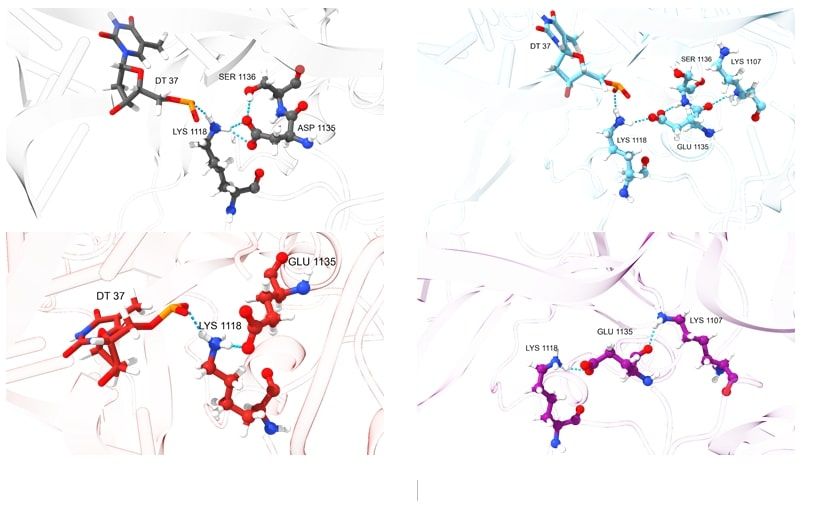
It is intriguing that in the wild-type Cas9, the aspartate at position 1135 forms hydrogen bonds with lysine at position 1118 and serine at position 1136. Lysine 1118, in turn, forms a bond with the phosphate of the DNA side chain due to its positively charged amino group, which is observed in the mutations D1135E and D1135E+L1206P. Mutations at positions 147 and 411 apparently distort the conformation of the Cas9 protein's side chain, preventing lysine from forming a hydrogen bond with the residue of DNA's phosphoric acid.
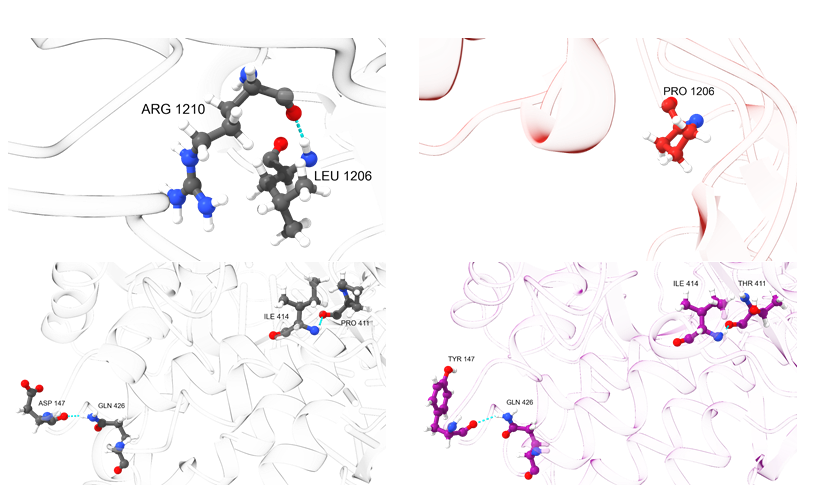
The replacement of leucine with proline in the mutation D1135E+L1206P leads to the inability to form a hydrogen bond with arginine at position 1210, which can be observed in the wild-type Cas9. At the same time, mutations at positions 147 and 411 do not change the nature of hydrogen bonds, although these particular changes lead to more profound conformational distortions along the polypeptide chain (based on the analysis of the geometry of the region of the 1135 amino acid).
Thus, thanks to MD simulations, it was found that the studied mutations are the source of conformational changes in the protein's tertiary structure, specifically in regions that are much farther from the amino acid substitutions (see Fig. 1), thereby revealing one of the reasons for the change in the activity of mutant forms relative to the wild-type form of Cas9.