Navigating the Virtual Lab: In Silico Analysis in Rational Drug Design
This article explores in silico methods crucial for rational drug design in pharmaceutical research. It discusses the integration of experimentalists and theoreticians, emphasizing the cost-effectiveness and predictive power of in silico methods in streamlining research.
Table of Contents
- Molecular dynamics (MD) simulations
- Quantum Chemical (Mechanical) studies
- QM/MM
- How to Strengthen Your Product with in silico Analysis?
- Our Solution
Few words about in silico in researcher's life
Development of any product, from drugs to protein vaccines, is a very complex and costly process. We can observe this in the development of pharmaceuticals: over the past decades, pharmacies have seen a large number of drugs with good therapeutic effects and tolerability. This has been the result of both the overall development of pharmacology and the implementation of in silico methods in particular. The screening and selection stage from thousands of candidate compounds cannot be carried out without in silico analysis. It is responsible for the exponential growth in the number of discovered compounds. Rational drug design has replaced empirical search, where chemists accidentally discovered a compound capable of producing a therapeutic effect, thereby accelerating and simplifying drug development. This acceleration is due to the fact that in silico analysis methods allow virtual reproduction of the key chemical properties of a substance based on its assumed atomic structure. It is these methods that I would like to talk about.
Few words about the author
For me, Stan Bachurin, the topic of rational drug design is vital, as I have devoted more than half of my life to various aspects of this issue. I graduated as a pharmacist, completing the pharmaceutical faculty, where I studied pharmacology for 2 years and 9 sections of chemistry exploring different aspects of drug development, synthesis, and analysis. During my studies, I became familiar with SAR (Structure-Activity Relationship) and QSAR (Quantitative SAR) analysis of drugs. After graduation, I lectured to students on pharmaceutical and toxicological chemistry courses while simultaneously working on Ph.D. theses. During my years in graduate school, I became fascinated with in silico methods and have since been unable to imagine my life without them. After obtaining my Ph.D., I became interested in applying in silico methods to solve biochemical problems and began teaching courses in bioorganic chemistry and biochemistry. I did not focus solely on one calculation method but tried to cover all possible methods applicable to the study of protein, membrane, nucleic acid, and small molecule properties.
Many researchers, who lead pharmaceutical companies, may have a good scientific background and an idea of developing a drug substance. Often, these are experimentalists, as their character traits make them full of determination and courage to materialize their ideas regardless of the difficulties they may face. Experimentalists are risk-takers. Theoreticians, as I can judge from myself and many colleagues, are cautious individuals who feel comfortable working knowing that any mistake can be corrected. Therefore, those who are bold tend to gravitate towards flasks, while those overly cautious tend towards computers. The dream team, of course, is to have both an experimental department and a theoretical department. However, this is not always feasible, as project budgets are limited, and cost-saving measures are necessary where possible. At the same time, in silico methods are needed precisely to peek a few steps ahead and eliminate obviously futile research paths or discover new ones that were previously not obvious, which can save a money and time on experiments. Plus, in silico analysis results can be presented in a colorful enough light to convince an investor of the high potential of the project.
In this article, we will explore the key in silico analysis methods used in rational drug development and, in conclusion, discuss how our solution can save your business time and money.
For comprehensive analysis, three main calculation methods are sufficient: Quantum Mechanical (QM), Molecular Dynamics (MD) simulation, and Quantum Mechanical/Molecular Mechanical (QM/MM). All the key information necessary for further work can be obtained in a very short time after processing the results of calculations using these methods. It is impossible to cover all possible analysis methods in one article, and there is no need to do so, as different approaches are required for different tasks. However, the key trio will always remain unchanged.
1. MD SIMULATIONS
Molecular Dynamics (MD) simulation is a method used to study macromolecules such as proteins, nucleic acid fragments, membrane structures, and their combinations. In this method, each atom is represented by a charged sphere on a spring, where the spring simulates the chemical bond. To perform calculations, a file with force fields is required, which is a table containing all the necessary information for modeling the molecule: the sizes and masses of each type of atom, the bond lengths between each pair of atoms, as well as the values of planar and dihedral angles between corresponding atoms. For each geometric parameter, additional energy barriers are specified. Many force fields also specify van der Waals radii. Obviously, for example, carbon atoms depending on their position will have different bond lengths, so force fields may contain several types of carbon atoms. Force fields are created for a specific class of compounds – proteins, carbohydrates, etc., although there are also universal ones – GAFF2, UFF, etc.
All values for force fields are taken from series of accurate quantum chemical calculations and experiments, so force fields are often updated, and many research groups develop their own versions. If you look at what they are, you will understand how difficult it can be to choose the right one for your research. The choice of force fields depends on the task at hand, the researcher's experience, and preferences.

MD simulation is necessary for preparing targets for the investigated drug substance. On the rcsb.org or Uniprot websites, you can obtain an atomistic model of the protein of interest that interacts with the developed drug substance. Most of these models are obtained by X-ray crystallography, and the crystalline structure of the substance differs significantly from the dissolved one. To obtain a more adequate model, MD simulation is applied. Within the scale of several thousand atoms, quantum effects can be neglected, and practice shows that MD simulations in force fields are quite adequate for such tasks. The researcher needs to set key conditions – the size of the system (boxes) in which the simulation is performed, the type and number of ions, temperature, pressure, simulation time, etc. Sometimes additional interventions are required, such as adding special ions to the active center, etc. Quality MD simulations are multi-stage, as artifacts and errors can occur during system preparation at the stages of optimizing the initial geometry and equilibration. There are no universal tips and approaches here, only the experience and chemical intuition of the theorist. As a result of this stage, we obtain not only a reasonable and ready-to-use geometry of the target for our future drug but also a lot of valuable information about it: conformational rigidity, dynamics of hydrogen bond formation/breakage during simulation time, changes in sizes in dynamics, etc. Moreover, after obtaining results and data on the drug-binding domain, you can raise the results again and quickly analyze the same conformational lability, hydrogen bond rearrangements, etc., but already for a separate domain. The main software packages for MD simulation are GROMACS, AMBER, LAMMPS, CHARMM, CP2K, etc.

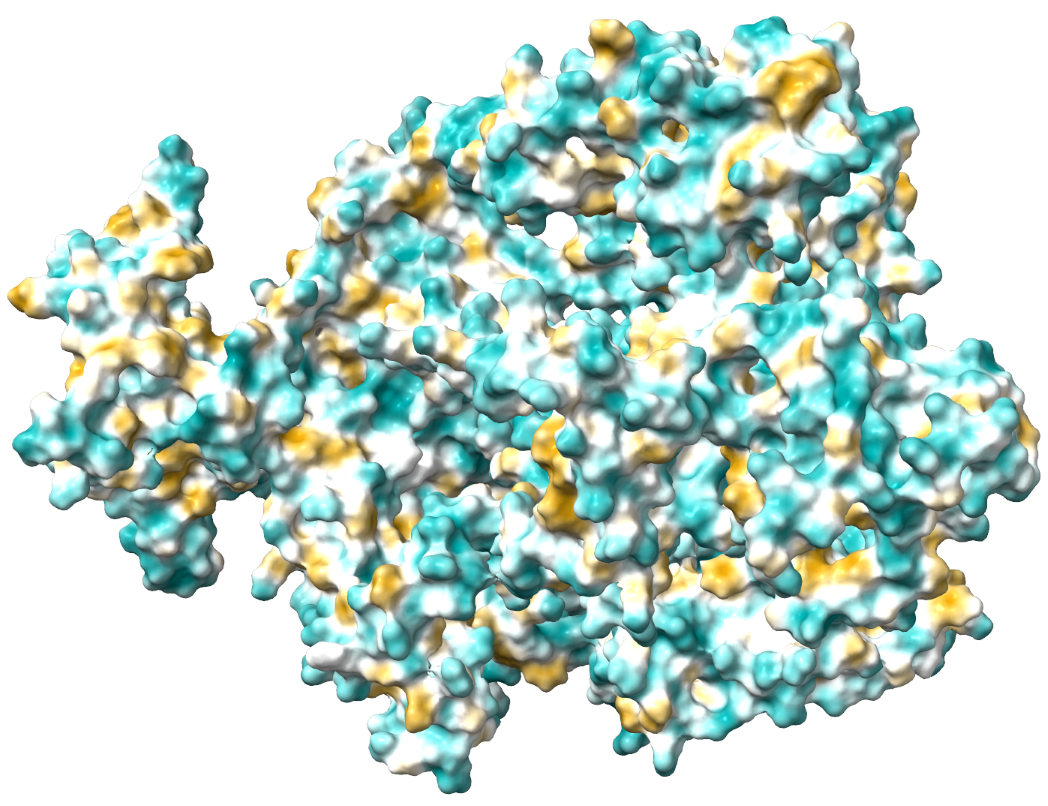
2. Quantum Chemical (Mechanical) studies
How do we search for new substances? Each subsequent generation of drugs should be more effective and safer than the previous one. Before the advent of in silico methods, there was a whole branch of chemistry that fortunately became history - combinatorial chemistry. Back then, a large number of experimental animals were used, and dozens of structures with minor structural differences were tested on them. Rarely did any positive effects emerge even on one group. More often than not, the efforts of synthetics and experimenters yielded no results. QM and its further development into QM/MM methods made drug development more rational. We'll talk about QM/MM in the next section, but here let's briefly discuss QM analysis methods.
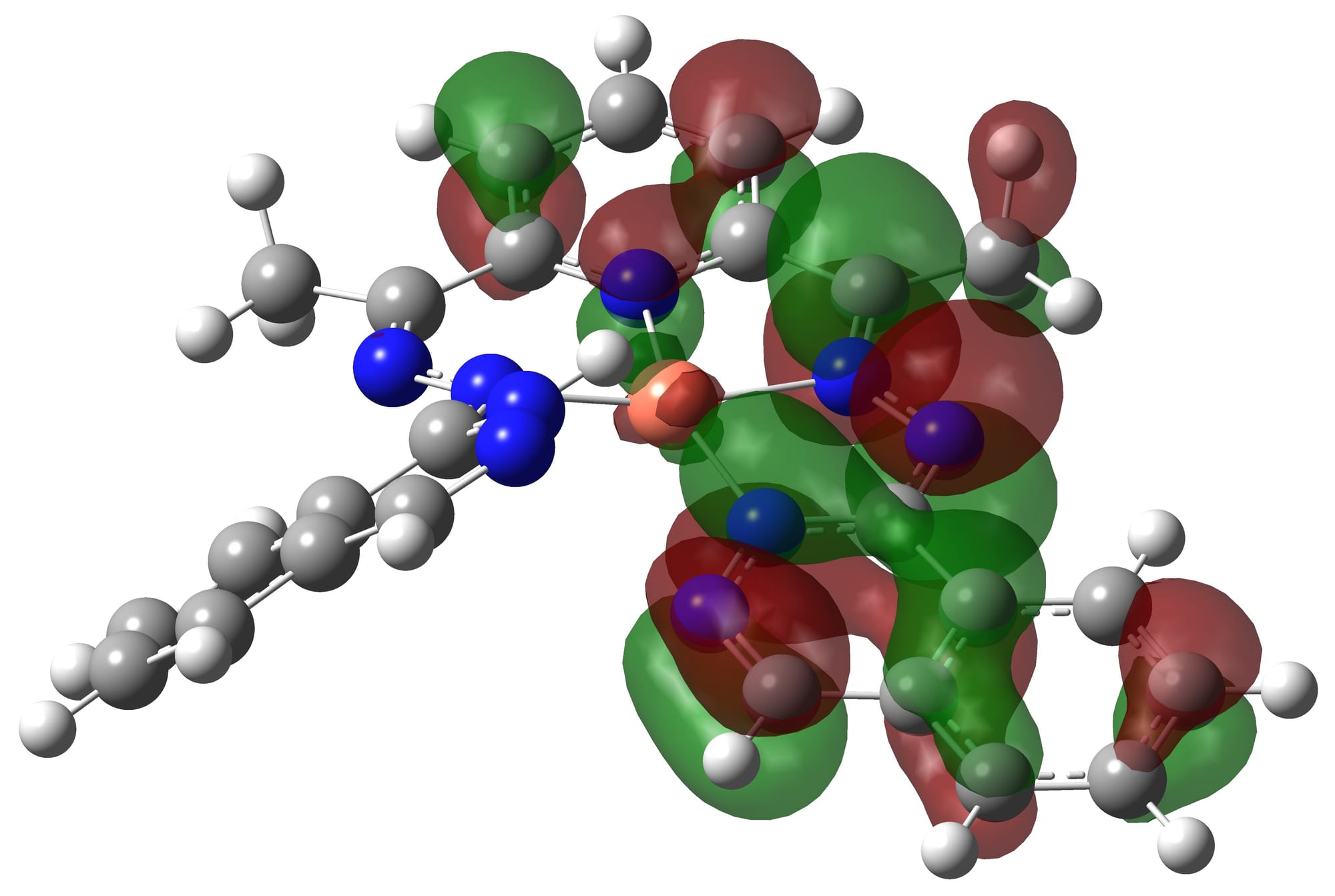
QM methods allow us to obtain a set of important data about low-molecular-weight substances – optimal geometry, charge distribution, volume, structure of valence molecular orbitals, which is necessary for explaining reactivity. All of this is achieved by solving the Schrödinger equation. There are many methods that use some tricks to reduce computational cost. There are very cheap semi-empirical methods (pm7, am1, pm6mm), there are very expensive and very accurate ab initio methods – Coupled clusters, MP2, MP4, etc., but the golden mean is DFT methods, including the recently developed DFTB methods. In addition to choosing the method, the researcher needs to select the functional and basis set in which to conduct QM calculations. Here we will not delve into these concepts in detail, as they require a separate discussion. Let's just say that the choice of functional and basis set determines the accuracy and correctness of the calculation results, and if an incorrect basis set and functional are selected for the system, the results will be unsuitable for research. At the dawn of the work, the theorist had to rearrange computational experiments and rewrite the results due to inexperience.
At this stage, we construct various variants, from which candidates for further research will be selected. It is desirable to model several (2-3) reference structures, i.e., existing analogs, so that based on in silico analyses for them, we can assess the potential of future drugs. Here, I want to make a brief digression and say a few words about bioisomerism – an extremely important concept for rational drug design, which is not always mentioned.
Bioisomerism
In the chemistry course, isomers are discussed as compounds with the same quantitative-qualitative atomic composition but different structures. Given the variety of differences in structure, several types of isomerism are distinguished – geometric, optical, structural, etc. However, bioisomerism is a completely separate type of isomerism. Bioisomer substances can differ greatly in structure and consist of completely different atoms while having similar biological effects. The reason for the similarity of biological action is their ability to bind in the same domain of the target molecule (enzyme, receptor, nucleic acid, etc.). This is because bioisomers have the same charge and key distances (you can compare the structure of diethylstilbestrol and estrone) at binding points with amino acid residues in the target, allowing them to produce similar effects (or, conversely, to switch off the effects of their bioisomer by occupying the target domain without further development of events).
Based on QM calculations, we can obtain custom force field parameters for our candidates to use them in subsequent in silico analysis. For QM calculations, packages such as Gaussian, CP2K, GAMES, Quantum Espresso, etc., are used. The author began his work in Gaussian 09 and became acquainted with the world of quantum chemistry thanks to the simplicity of working with this package. However, gaining experience, he switched to CP2K, which is less user-friendly in terms of writing calculation protocols but allows controlling every aspect of the calculations, which is enticing.
3. QM/MM
Before proceeding directly to QM/MM analysis, we need to determine the binding sites of our ligand with the target. This can be done through docking analysis in specially designed programs or by conducting MD simulation, where the target and several candidate drug molecules are present. In most cases, both approaches will yield the same ligand-target complex structure. We will work with a set of such ligand-target complexes.
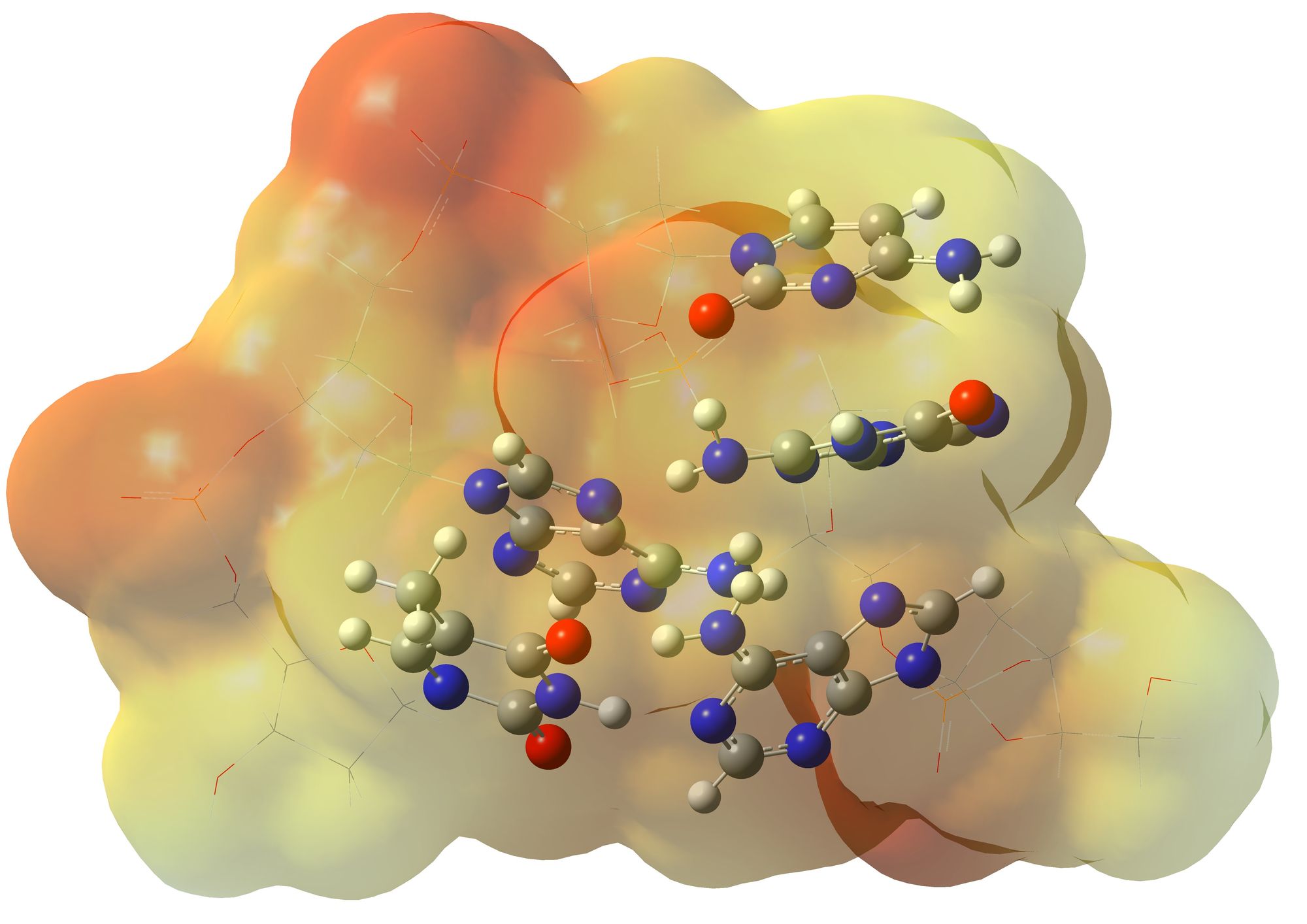
Thanks to the QM/MM approach, we can assess how well a substance binds to the receptor. To do this, we specify to the program, in which we will perform the calculations, which amino acid residues of the target protein form the ligand-binding domain, so that the package calculates them and the ligand using QM methods, while everything else (solvent, backbone of the target protein, etc.) is calculated using MM methods. Then, for each complex, we calculate the energies of three systems: the target, the target-ligand complex, and the ligand (if the functional and basis set for QM are the same as those used in the second phase of the study, then essentially we already have this energy value). We determine the binding energy by the difference between the energy of the target-ligand complex and the sum of the energies of the target and the ligand separately. Then, we compare the energies of the obtained complexes with the references and draw conclusions. Modifications with higher binding energy are potential candidates for the next generation of studied drugs, and it makes sense to materialize them. It is desirable to select several such candidates because in subsequent experiments, unforeseen side effects may occur, which are difficult to predict. If the original substance has side effects, we can model its interaction with another receptor responsible for the side effect in the same way, but now we need to catch a structure with lower binding energy. This is, in general terms, how in silico methods can significantly narrow down the search and save time and money on experiment optimization. Moreover, even beyond these in silico methods, we can obtain the charge distribution in the binding domain, analyze it, and propose new candidates for drug substances with more congruent geometry and charge distribution on the surface. For QM/MM calculations, CP2K is better suited as it works well with external force fields.
How to Strengthen Your Product with in silico Analysis?
In this article, we briefly overviewed in silico methods and their applications. In silico is a distinct branch of scientific research with a rich arsenal of tools. Essentially, the combination of various methods allows for modeling any task and solving it. If you feel that in silico analysis can benefit your project, there are several ways to proceed:
- Self-Study of In Silico Methods: You need to become proficient in the Linux command line, as most packages are optimized for this operating system. Choose the most suitable distribution where you won't encounter problems with installing necessary packages such as Python, BLAS and LAPACK, cuFFT, various compilers for Fortran, C, and C++, etc. You'll need to acquire or rent a powerful computer since computations require substantial resources if you want quick results. Setting up and optimizing the necessary packages for computations is another challenge. Expect to spend a lot of time on learning and configuration, and be prepared for false positive and false negative results initially due to incorrect initial settings in computation protocols. In any case, you'll have to practice on classical systems.
- Consulting Experts: Seek assistance from specialists in in silico analysis. They can provide the necessary expertise and guidance. This can be through individual consultations or collaboration with a team of researchers.
- Collaboration with Institutions or Laboratories: Collaborate with an institution or laboratory specializing in this field. This option is the most reliable but also the most expensive and time-consuming. Institutions may have limited staff and busy schedules due to grants. You'll need to formalize an agreement and communicate with the laboratory through official channels, which may not always be convenient.
- Engaging a Firm Specializing in Such Analyses: Find a company that specializes in these calculations. This option is more attractive than the previous one, as negative aspects such as communication barriers and execution times will be minimized. However, it's essential to carefully choose whom to entrust with in silico analysis.
If you're commissioning calculations and don't have a theoretical chemist on staff, those you work with should provide in silico analysis results in both raw (output files directly for independent review by other researchers) and processed forms, along with their conclusions. The conclusions should be written in a way that you fully understand the obtained result.
Contact us for a free consultation to discuss your issue
Book a Call
Our solution
In silico methods are not something otherworldly, and the interpretation of results should be accessible to a wide range of researchers. A few years ago, biotechnologists contacted us by chance, asking to conduct structure calculations for various mutants of an enzyme. As a result of the communication, it became clear that they simply didn't know about in silico methods. Therefore, we created LambasLab to help scientific and technical businesses use in silico methods to develop their products and enrich humanity with new high-quality products. We addressed several organizational problems of working with theoretical chemists thanks to the experience of our employees in this field:
- We solved the problem of computational resources by writing our Docker containers for computational packages and renting servers with graphics cards. This allows us to easily scale for almost any task.
- Our team consists of specialists in biochemistry, so we focus on solving biological problems—from calculations necessary for drug development to modeling biochemical processes.
- We have developed our own system of protocols for in silico calculations, which saves time in project preparation and ongoing organization of computations. It also makes the work reproducible if you need to return to experiments with a different set of candidate substances.
- We easily guarantee confidentiality. It's important to understand that in silico methods are a complex scientific field that provides answers in the form of energy, geometry, dynamics of structure changes, etc. Without a specific background in the task, a theoretical chemist cannot implement or convey the results—they are meaningless without context.
Request calculations with clear, basic result outputs in the form of corresponding graphs and values. Prepare a list of questions you would like answered. Our theoreticians will address specific questions without delving into unnecessary details.